
En collaboration avec différents partenaires de recherche, les équipes de DeepMind viennent de lâcher une véritable bombe scientifique, assez inattendue, dont les retombées pourraient bien redéfinir la biologie moléculaire. Grâce à la dernière version de leur IA baptisée AlphaFold, les chercheurs ont produit une base de données colossale, aujourd’hui disponible en accès libre, qui recense la structure 3D de presque toutes les protéines présentes dans le corps humain.
La portée de ces travaux est phénoménale; c’est une avancée que certains scientifiques de haut vol n’hésitent pas à mettre dans le même sac que le séquençage du génome de notre espèce. Une comparaison osée, quand on sait à quel point cette étape avait catapulté la recherche médicale en avant de plusieurs années, mais néanmoins parfaitement justifiée.
“C’est le type d’avancée qu’on voit une fois par génération”
– Arthur D. Levinson
Nous vous proposons donc un tour d’horizon de ces travaux qui révolutionnent déjà la science des protéines.
Pourquoi est-ce important ?
Les protéines, ce sont les éléments à partir desquels est construite la vie. Elles travaillent de concert dans un ballet infiniment complexe, mais parfaitement coordonné, qui conditionne tout ce qui se passe au sein de notre métabolisme. C’est grâce à elles que nous pensons, que nous bougeons, que vous voyons… que nous existons, en somme. Leur étude est donc d’une importance capitale dans de très nombreux domaines de la médecine. Ce que nous savons du fonctionnement de notre corps, nous le devons en grande partie à l’étude de ces objets.
Quand certaines protéines viennent à manquer, cela peut générer un tas de pathologies graves. Mais c’est aussi vrai lorsqu’elles sont endommagées, ou repliées de façon incorrecte (voir ci-dessous); elles deviennent alors incompatibles avec certains autres acteurs de notre organisme, avec toutes les conséquences que cela implique. À bien des égards, connaître la structure 3D des protéines est donc un avantage considérable.
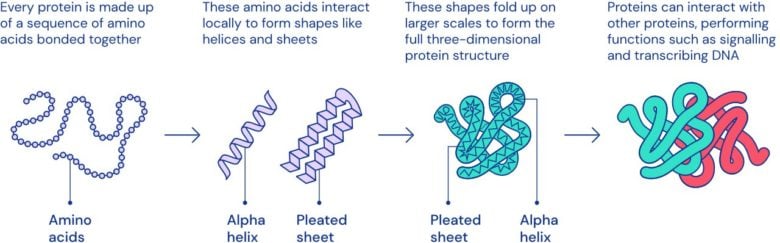
Pourquoi est-ce si difficile de les étudier ?
Puisque ces protéines sont si importantes, on peut se demander pourquoi ce travail n’a pas déjà été réalisé. Après tout, le séquençage du génome auquel cette base de données a été comparée a été réalisé dès 2003. Mais aussi complexes soient-ils, ces travaux seraient presque simples par rapport à ceux qui s’attaquant à la cartographie des protéines.
L’ADN, c’est un assemblage d’un petit nombre d’éléments différents, empaquetés dans un emballage moléculaire bien spécifique; si l’on raisonne au niveau le plus basique, l’immense complexité de notre patrimoine génétique tient donc aux innombrables combinaisons d’à peine quatre éléments différents dans l’ADN. En revanche, les protéines qui nous composent sont construites à partir de… 20 acides aminés (ou plus précisément, de leurs résidus). Cela démultiplie les possibilités et complexifie grandement leur analyse.
Mais ce n’est que la partie émergée de l’iceberg. Comme indiqué plus haut, la fonction des protéines ne dépend pas seulement de l’ordre dans lequel sont disposés les éléments; chacune est repliée sur elle-même dans une structure en trois dimensions qui lui est propre et conditionne entièrement son fonctionnement. Cela rajoute une couche de complexité extraordinaire à leur analyse.
Si l’ADN était une sorte de télégramme, c’est à dire un message de forme assez simple et très codifié, le ballet des protéines serait un vrai langage complet, avec ses nuances, sa ponctuation et ses figures de style qui peuvent radicalement changer le sens d’une phrase. Vous connaissez certainement l’exemple célèbre, “On mange, les enfants !”; il suffit de supprimer la virgule ou de changer de ton pour passer d’une invitation routinière à un aveu de cannibalisme infantile. Pour comprendre la différence entre les deux phrases, lire la succession des lettres ne suffit pas. Il faut comprendre les deux parties de la phrase indépendamment, et le lien fonctionnel qui les unit.
Lorsqu’on souhaite étudier une protéine, c’est la même chose : s’intéresser à la succession des composants ne suffit pas. Il faut aussi découper l’ensemble en plusieurs parties avec chacune sa fonction propre, un peu comme on chercherait le sujet ou le complément d’une phrase. Il faut ensuite observer la façon dont ces différents domaines interagissent (la grammaire, la conjugaison…), pour pouvoir enfin comprendre la structure de la protéine et son rôle (le sens de la phrase, en somme).

Traditionnellement, c’est un problème qui a mis les plus grands esprits de la discipline à rude épreuve. Ces analyses sont réalisées de façon plus ou moins empirique; le processus est effroyablement long et peu efficient. Il peut souvent prendre des mois pour une seule protéine – et sans garantie d’avoir un résultat exploitable ! Les chercheurs ont donc fait appel à l’informatique pour modéliser ces phénomènes. Une approche a permis de faire des progrès considérables et de prédire la structure 3D de nombreuses protéines. Mais le problème est si complexe que les algorithmes basés sur la force brute ont rapidement atteint leurs limites. Après des décennies d’études acharnées, les chercheurs disposaient d’informations structurelles solides sur à peine 17% du protéome humain.
Qu’est-ce qui a changé ?
C’est là qu’intervient DeepMind, une société satellite de Google spécialisée dans la recherche de pointe en intelligence artificielle. En 2019, l’entreprise a jeté un énorme pavé dans la mare en proposant une approche révolutionnaire, basée sur le deep learning. Leur programme AlphaFold a réussi à prédire la structure 3D de protéines à partir de leur composition, avec des temps de calcul ridiculement courts par rapport aux méthodes d’analyse standard et une excellente précision.
La communauté scientifique a tout juste eu le temps de s’en émouvoir que la même équipe revenait en 2020 avec une preuve de concept d’une rapidité phénoménale, qui a battu tous les systèmes prédictifs créés depuis que l’idée a germé à la fin des années 90; de quoi méduser les chercheurs actifs dans cette discipline.
“Ce qui nous prenait des mois ou des années, AlphaFold l’a fait en un week-end”
– John McGeehan
Partout, les réactions se multiplient pour saluer cette avancée sur un problème vieux de presque 50 ans. De l’aveu d’Andrei Lupas, biologiste réputé du Max Planck Institute for Developmental Biology, cette technique lui a déjà permis de dompter en deux temps, trois mouvements une protéine qui menait la vie dure à son laboratoire depuis… plus de dix ans.
“Ça va changer la médecine. Ça va changer la recherche. Ça va changer la bio-ingénierie. Ça va tout changer”.
-Andrei Lupas
C’est ce même système qui a permis aux équipes de DeepMind de constituer ce gigantesque catalogue d’origami moléculaire pour bio-ingénieurs. Ils ont simplement appliqué leur technique à des dizaines de milliers de protéines, dont la quasi-totalité de celles de notre organisme. Les chercheurs du monde entier peuvent désormais visualiser le repliement en 3D de 98.5% des protéines attendues du corps humain. Et surtout, ce qui leur prenait auparavant des mois est désormais disponible en deux clics.
Qu’est-ce que ça change aujourd’hui ?
Certes, toute cette base de donnée n’est pas entièrement exploitable en l’état. Mais méthodologiquement parlant, cela représente un changement de paradigme si radical qu’il pourrait perturber les habitudes des chercheurs. C’est en tout cas l’avis de la généticienne réputée Edith Heard, citée par TechCrunch. “Les biologistes structuraux ne sont pas encore habitués à l’idée qu’ils peuvent simplement vérifier n’importe quoi en quelques secondes, plutôt que de prendre des années pour vérifier expérimentalement”, explique celle qui est aussi directrice du prestigieux Laboratoire européen de biologie moléculaire.
Mais la recherche fondamentale ne sera pas la seule à bénéficier de cette base de données. Médicalement parlant, elle pourrait avoir des implications assez rapidement. C’est notamment le cas pour toutes les maladies que l’on suspecte d’être causées par des protéines non fonctionnelles. Par exemple, pour identifier les causes probables d’une maladie génétique, il aurait auparavant fallu des mois, voire des années pour tester une seule protéine; avec toutes ces informations à portée de clic, il sera parfois possible d’arriver à la phase concrète beaucoup, beaucoup plus rapidement.
C’est encore plus important pour les maladies orphelines rares. Bien souvent, la recherche est très longue à cause de leur faible représentation. Maintenant que le processus pourrait être largement facilité, cela offre de nouveaux espoirs de développer des traitements pour des pathologies parfois uniques.

Quel avenir pour AlphaFold ?
Une fois les chercheurs habitués à ce mode de fonctionnement, ces travaux pourraient ouvrir la voie à d’autres projets à l’ambition encore plus démesurée. Certaines questions qui n’avaient même pas de sens pratique auparavant pourraient désormais être mises sur la table.
Un exemple : puisqu’analyser une seule protéine prenait autant de temps, il était tout simplement inutile d’espérer simuler leurs interactions à grande échelle au sein d’un système exponentiellement plus complexe. Informatiquement parlant, nous ne disposions tout simplement pas de la technologie nécessaire pour arriver à des résultats exploitables. Mais les performances d’AlphaFold montrent que grâce au machine learning, ces questions qui relevaient autrefois de l’expérience de pensée pourraient devenir une réalité, avec tout ce que cela implique en termes de progrès scientifique. Pour Arthur D. Levinson, fondateur de Genetech, l’une des entreprises associées au projet, “cela montre comment les méthodes calculatoires sont parties pour transformer la recherche en biologie, et sont pleines de promesses pour accélérer le processus de découverte”.
Et c’est bien là le plus intéressant. C’est difficile à concevoir, tant il s’agit déjà d’une avancée conséquente, mais AlphaFold n’est que le produit précoce d’une niche technologique encore balbutiante. Si elle ne fait que résoudre un problème très précis, elle va sans aucun doute ouvrir un nouvel horizon de recherche pour tout le futur de la bio-ingénierie calculatoire. Une vraie petite révolution.
Documents annexes :
Le texte de l’étude est disponible ici.
Le communiqué de DeepMind est disponible ici.
Les travaux précédents de DeepMind sur AlphaFold sont disponibles ici.
La base de données est disponible ici.
🟣 Pour ne manquer aucune news sur le Journal du Geek, suivez-nous sur Google et sur notre canal WhatsApp. Et si vous nous adorez, on a une newsletter tous les matins.











Un bel article, bien écrit et fort intéressant. Bravo M. Gautherie ! Merci pour la clarté des sources.
Article très bien écrit ça fait plaisir de voir un article d’une telle qualité sur ce site. Merci d’avoir ajouté les sources également.
Je plussoie mes camarades de lecture ! Bravo
Super article!
Je me joins aux autres commentaires pour saluer la prestation journalistique. Merci, enfin un article bien écrit, structuré, simple à comprendre et sans fautes d’orthographe…
Super lecture
Merci
Article didactique très clair, sans faute et sourcé: mes compliments à mon tour au journaliste et à la rédaction.